Definition
Mitochondrial disease result from failures of the mitochondria, specialized compartments present in every cell of the body (except red blood cells). Mitochondria are responsible for creating more than 90% of the energy needed by the body to sustain life and support organ function. When they fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail.
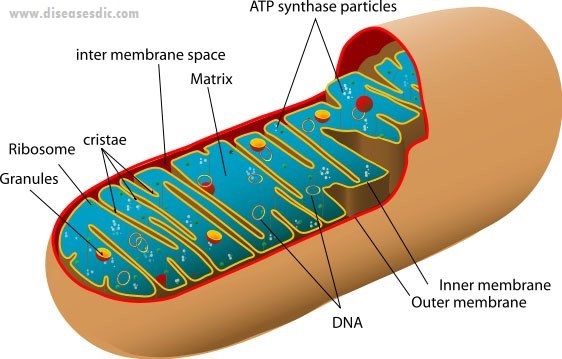
Human mitochondrion
The parts of the body, such as the heart, brain, muscles, and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected. Although mitochondrial disease primarily affects children, adult onset is becoming more common.
Epidemiology
Infants, children, and adults may develop mitochondrial disorders. Experts in mitochondrial medicine describe a spectrum of disease, ranging from mild to severe. 1 in 4,000 people are estimated to have a genetically confirmed primary mitochondrial disease, yet many remain undiagnosed.
In adults, many diseases of aging have been found to have defects of mitochondrial function, including, but not limited to, diabetes, Parkinson’s disease, Huntington’s disease, atherosclerotic heart disease, stroke, Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), autoimmune disorders, environmental toxicities, and cancer.
Who is at risk?
The more energy a cell needs, the more mitochondria they have. Because our brain, heart, liver, kidneys, digestive tract and muscles need the most energy, they are the most susceptible to mitochondrial disease.
Causes and it types
In most people, primary mitochondrial disease is a genetic condition that can be inherited (passed from parents to their children) in several ways.
To understand inheritance types, it’s helpful to learn more about genes and DNA. Genes are substances that give us our traits, such as brown eyes or blue eyes. Genes contain DNA, which is the “blueprint” that gives each person his or her unique makeup.
Under normal circumstances, a child inherits genes in pairs one gene from the mother and one from the father. A child with a mitochondrial disease does NOT receive a normal pair of genes from the parents. The gene has mutated – meaning it has become defective (changed). Learning the way a mitochondrial disease has been inherited helps predict the chance of passing on the disease(s) to future children.
Inheritance types are:
Autosomal recessive inheritance: This child receives one mutated copy of a gene from each parent. There is a 25% chance that each child in the family will inherit a mitochondrial disease.
Autosomal dominant inheritance: This child receives one mutated copy of a gene from either parent. There is a 50% chance that each child in the family will inherit a mitochondrial disease.
Mitochondrial inheritance: In this unique type of inheritance, the mitochondria contain their own DNA. Only mitochondrial disorders caused by mutations in the mitochondrial DNA are exclusively inherited from mothers. If this is the way a mitochondrial disease was inherited, there is a 100% chance that each child in the family will inherit a mitochondrial disease.
Random mutations: Sometimes genes develop a mutation of their own that is not inherited from a parent.
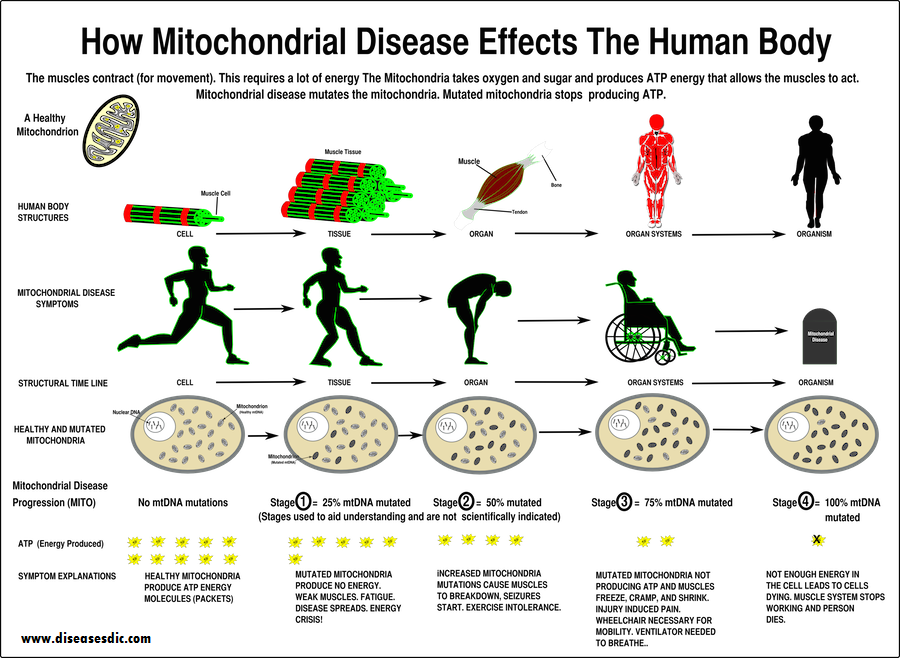
Symptoms of mitochondrial disease
The severity of mitochondrial disease symptoms is different from person to person. The most common symptoms are:
- Poor growth and failure to thrive (in children)
- Loss of muscle coordination, muscle weakness, and pain, low tone, exercise intolerance • Neurological problems, seizures
- Autism, autistic spectrum, autism-like features
- Visual and/or hearing problems
- Developmental delays, learning disabilities
- Movement disorders
- Heart, liver or kidney disease
- Gastrointestinal disorders, including severe constipation, diarrhea, swallowing difficulty, repeated vomiting, cramping, reflux
- Diabetes
- Increased risk of infection
- Neurological issues, including difficult to treat seizures, migraines, and stroke or stroke like events
- Thyroid and/or adrenal dysfunction
- Autonomic dysfunction (may affect the functioning of the heart, bladder, intestines, sweat glands, pupils, and blood vessels
- Respiratory issues
- Lactic acidosis (the buildup of lactate in the body, which results in an excessively low pH in the bloodstream)
- Neuropsychological changes characterized by confusion, disorientation, dementia, and memory loss
Diagnosis and test
Unfortunately, mitochondrial genetic disorders can be difficult to diagnose, and many affected people may never receive a specific diagnosis. They are often suspected in people who have a condition that effects multiple, unrelated systems of the body. In some cases, the pattern of symptoms may be suggestive of a specific mitochondrial condition. If the disease-causing gene(s) associated with the particular condition is known, the diagnosis can then be confirmed with genetic testing.
If a mitochondrial genetic disorder is suspected but the signs and symptoms do not suggest a specific diagnosis, a more extensive work-up may be required. In these cases, a physician may start by evaluating the levels of certain substances in a sample of blood or cerebrospinal fluid. Other tests that can support a diagnosis include:
- Exercise testing
- Magnetic resonance spectroscopy (detects abnormalities in the brain’s chemical makeup)
- Imaging studies of the brain such as MRI or CT scan
- Electroencephalography (EEG)
- Tests that evaluate the heart including electrocardiography and echocardiography
- Muscle biopsy
When possible, confirming a diagnosis with genetic testing can have important implications for family members. Identifying the disease-causing gene(s) will give the family information about the inheritance pattern and the risk to other family members. It will also allow other at-risk family members to undergo genetic testing.
Mitochondrial disease treatment
There are no cures for mitochondrial diseases, but treatment can help reduce symptoms or slow the decline in health.
Treatment varies from patient to patient and depends on the specific mitochondrial disease diagnosed and its severity. However, there is no way to predict a patient’s response to treatment or predict how the disease will affect that person in the long run. No two people will respond to the same treatment in the same way, even if they have the same disease.
Treatments for the mitochondrial disease may include:
Vitamins and supplements, including Coenzyme Q10; B complex vitamins, especially thiamine (B1) and riboflavin (B2); Alpha lipoic acid; L-carnitine (Carnitor); Creatine; and L-Arginine.
Exercises, including both endurance exercises and resistance/strength training. These are done to increase muscle size and strength. Endurance exercises include walking, running, swimming, dancing, cycling and others. Resistance/strength training include exercises such as sit-ups, arm curls, knee extensions, weight lifting and others.
Conserving energy: Don’t try to do too much in a short period of time. Pace yourself.
Other treatments: These may include speech therapy, physical therapy, respiratory therapy, and occupational therapy.
Avoid situations that can make your medical condition worse. These include: exposure to cold and/or heat; starvation; lack of sleep; stressful situations; and use of alcohol, cigarettes and monosodium glutamate (MSG, a flavor enhancer commonly added to Chinese food, canned vegetables, soups, and processed meats).
Prevention of Mitochondrial disease
- The transmission of mitochondrial disease is dependent on the proportion of mutated mtDNA carried by the affected mother’s egg. Homoplasmic mutations will be transmitted to each child in a homoplasmic state.
- However, in women with heteroplasmic mutation loads, the level transmitted to their children is more difficult to predict. The only methods to avoid transmission of mutated mtDNA are those dependent on egg donation, including mitochondrial donation where there remains a small risk of mutation carryover.
- Other methods such as PGD are risk reduction procedures and are unlikely to completely prevent transmission of the mutated mtDNA. Preimplantation genetic diagnosis and/or prenatal diagnosis are offered to women who have heteroplasmic mtDNA mutations but are aiming to use their own eggs to have a baby and women having a mitochondrial disease due to a nuclear mutation.
- A preimplantation genetic diagnosis may be offered as part of an in vitro fertilization (IVF) cycle prior to the pregnancy. Prenatal diagnosis will be carried out when the pregnancy has been established for some time (chorionic villus sampling at 11-12 weeks’ gestation or amniocentesis at 15-17 weeks).
- However, none of these techniques can be offered to women with homoplasmic mutations. Mitochondrial donation is a new approach to prevent the transmission of mutated mtDNA from mother to child.
- The pronuclei, containing the nDNA of both parents, are removed from the fertilized egg and placed into a donor egg containing healthy mtDNA and from which the pronuclei have been removed, a technique known as pronuclear transfer. Alternatively, this IVF technique can be performed before fertilization using maternal spindle transfer and this may be ethically more acceptable to some couples.




exactly