Definition
Xeroderma pigmentosum or XP, is a very rare inherited disease that causes extreme sensitivity to the sun’s ultraviolet rays. Unless patients with XP are protected from sunlight, their skin and eyes may be severely damaged. This damage may lead to cancers of the skin and eyes. XP has been identified in people in every ethnic group all over the world.
Many persons with XP will get an unusually severe sunburn after a short sun exposure. The sunburn will last much longer than expected, perhaps for several weeks. This type of sunburn will usually occur during a child’s first sun exposure, and it may be a clue to the diagnosis of XP. However, some people with XP will not get a sunburn more easily than others, and the disease will be undetected until unusual skin changes appear over time.
 Xeroderma pigmentosum
Xeroderma pigmentosum
Abnormalities that occur in individuals with XPA, but are unlikely to be related to UV damage, include neurological abnormalities and internal cancers. At least 25 to 30% of individuals with XPA have progressive neurological issues that may include hearing loss, difficulty swallowing and talking, movement problems, seizures, and intellectual disability. The risk for internal cancers is thought to be linked to environmental carcinogens, like cigarette smoke and other, potentially uncontrollable, exposures.
History
Dermatologist Moriz Kaposi originally reported XP in 1874 after observing wrinkling, checkered pigmentation, tiny dilatations of the arteries, skin contraction, and the development of skin-based malignancies in four individuals with thin, dry skin. Following reports, the disease’s spectrum was enlarged to encompass neurologic problems, as well as a severe variant with dwarfism, gonadal hypoplasia, and learning disabilities, in addition to the classic symptoms of XP. The disease pathogenesis was identified as congenital excessive sensitization of the skin to ultraviolet radiation of the sun as early as 1926, with clear value in sun exposure prevention methods.
Epidemiology
XP has been found in all continents and across all racial groups. Consistent with autosomal recessive inheritance, males and females are similarly affected. Estimates made in the 1970s suggested an incidence in the USA of 1 in 250,000 and in Japan of 1 in 20,000. A more recent survey in Western Europe suggests approximately 2.3 per million live births. Anecdotally, the incidence in North Africa and the Middle East, where there is a high level of consanguinity, is substantially higher.
Symptoms of Xeroderma pigmentosum
Symptoms usually appear by the time a child is 2 years old.
Skin symptoms include:
- Sunburn that does not heal after just a little bit of sun exposure
- Blistering after just a little bit of sun exposure
- Spider-like blood vessels under the skin
- Patches of discolored skin that get worse, resembling severe aging
- Crusting of the skin
- Scaling of the skin
- Oozing raw skin surface
- Discomfort when being in bright light (photophobia)
- Skin cancer at a very young age (including melanoma, basal cell carcinoma, squamous cell carcinoma)
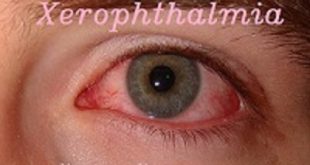
Eye symptoms include:
- Dry eye
- Clouding of the cornea
- Ulcers of the cornea
- Swelling or inflammation of the eyelids
- Cancer of eyelids, cornea or sclera
Nervous system (neurologic) symptoms, which develop in some children, include:
- Intellectual disability
- Delayed growth
- Loss of hearing
- Muscle weakness of the legs and arms
Xeroderma pigmentosum causes
Xeroderma pigmentosum is an autosomally recessive inherited disease, which means that a faulty xeroderma pigmentosum gene comes from each parent. Carriers of the xeroderma pigmentosum trait have one xeroderma pigmentosum gene and one normal gene and do not show signs or symptoms of the disease.
The signs and symptoms of xeroderma pigmentosum are a result of an impaired nucleotide excision repair (NER) system. Two types of NER have been identified:
- Genome (GG)-NER
- Transcription-coupled (TC)-NER.
At least seven different gene abnormalities or complementation groups have been described in different families (XPA to XPG) resulting in varying disease severity.
- XPA and XPC are relatively common
- XPE is fairly rare
- XPG is severe
- XPF is mild.
In addition to the genetic abnormality, the immunosuppressive effects of exposure to ultraviolet radiation (UV) contribute to the disease, for example by depleting Langerhans cells from the epidermis.
Xeroderma pigmentosum
Types of Xeroderma pigmentosum
There are 8 different genetic types of Xeroderma Pigmentosum depending on the affected gene: XPA, XPB (or ERCC3), XPC, XPD (or ERCC2), XPE (or DDB2), XPF (or ERCC4), XPG (ERCC5) and XPV (or POLH)10, each one of them affecting different mechanisms of DNA repair (making them more severe or not) in the protection of the aggressions caused by UV light.
| XP type | Affected gene / locus | Clinical features |
| XP-A | XPA / 9q22.3 | It represents 25% of the patients with XP. It is the most serious form of the disease because its DNA repair capacity is more diminished and its sensitivity to UV light highly increased. Patients present a diverse range of symptoms, including the development of numerous skin cancers at an early age and serious neurological abnormalities. |
| XP-B | XPB (ERCC3) / 2q21 | It is the least frequent of the XP types. It presents increased sensitivity to UV light. Patients may develop numerous skin cancers at an early age, some mild neurological abnormalities and they can present characteristics of Cockayne syndrome. |
| XP-C | XPC / 3p25 | It represents 25% of the patients with XP. It is considered the classical form of XP, the most frequent form in the Caucasian population. It is the disease variant with the highest capability to repair DNA even it shows increased sensitivity to UV exposure. Patients may develop severely atypical and dense lentigines at exposed areas as well as ocular anomalies but no neurological abnormalities. |
| XP-D | XPD (ERCC2) / 19q13.2-q13.3 | It represents 15% of the patients with XP. It presents increased sensitivity to UV light. Patients with this variant may develop numerous skin cancers at an early age. About neurological defects, they may present in some cases severe neurological defects with progressive degeneration such as sensorineural deafness, ataxia and mental retardation. |
| XP-E | XPE (DDB2) / 11p11-p12 ; 11q12-q13 | Low frequency. A less aggressive form of XP, it is limited to skin problems. Patients present the development of cutaneous cancers at a later age and they do not present neurological symptoms. |
| XP-F | XPF (ERCC4) / 16p13.3 | Low frequency. A less aggressive form of XP, most of the cases in the Japanese population. It presents the development of cutaneous cancers at a later age and it does not present neurological nor ocular symptoms. |
| XP-G | XPG (ERCC5) / 13q32-q33 | Low frequency but aggressive. Increased sensitivity to UV light. Development of numerous skin cancers at an early age and it presents neurological abnormalities in some cases. |
| XP-variant | XPV (POLH) / 6p21.1 | It represents 20% of the patients and it is very similar to XP-C. Increased sensitivity to UV exposure but less photosensitivity than other forms of XP. Higher risk of cutaneous cancers development after 30 years old and it does not present neurological symptoms nor ocular problems. |
Risk factors
XP is a hereditary disease. The only people at risk are those who have a parent, or parents, who either have the disease, or are carriers of the disease.
XP is more prevalent in certain isolated, geographic areas. This may, in part, be caused by consanguinity. This means that both parents are blood relatives, such as cousins. If parents share a genetic background, their chances of passing XP on to their children increase.
The most common complications of XP are skin cancer, neurologic abnormalities, and eye abnormalities.
Malignancies are also common. Repeated surgeries to remove tumors can result in disfigurement, but can be avoided by taking precautions against sun exposure.
Those with XP need to take extreme measures to protect every surface of the body from UV light at all times. These precautions include:
- wearing protective clothing, such as long sleeve tops, pants, and wide-brimmed hats
- applying broad spectrum sunscreen
- using UV-absorbing glasses with shields
Xeroderma pigmentosum Complications
There are many complications of xeroderma pigmentosum, which this activity discussed previously.
- The median age for the development of their first non-melanoma skin cancer is about 9. Patients may develop dozens to hundreds of non-melanoma skin cancers per year.
- Patients with xeroderma pigmentosum have a more than 10000 fold risk of developing non-melanoma skin cancer compared to the general population.
- The median age for the development of their first malignant melanoma is about 22.
- Patients with XP have a more than 2000 fold risk of developing malignant melanoma compared to the general population.
- The median age of death in patients with XP without neurodegeneration is about 37 years old. The median age of death in patients with XP with neurodegeneration is younger at about 29 years old.
- While the most common cause of death amongst patients with XP is metastatic malignant melanoma or invasive squamous cell carcinoma, the second most common cause is due to neurodegeneration.
Diagnosis
Clinical history and physical examination
In most cases, the initial clinical diagnosis can be made on the basis of extreme sensitivity to sun exposure, or by the appearance of freckles on the face at an unusually early age. A history of consanguinity between the parents may be an additional clue.
Confirmatory Tests for Xeroderma Pigmentosum
Tests to detect defective DNA repair
When there is a high index of suspicion, a piece of skin is removed (skin biopsy) using a local anesthetic and sent for a confirmatory test. These tests detect defective DNA repair and are offered in several countries.
A type of cells found in the skin termed fibroblasts that are hypersensitive when exposed to UV radiation can be studied with the help of a microscope to see how effective the DNA repair mechanism is. A blood sample can also be used to perform this test.
Histology
A skin biopsy may show findings of hyperkeratosis and increased pigmentation along with other findings which may confirm the diagnosis.
Genetic Testing
Molecular testing and gene sequencing can be done to identify the specific defect.
Prenatal diagnosis
Prenatal diagnosis may be done if there is already an affected sibling or one of the parents is affected by the condition. The DNA repair tests can be performed on amniotic cells or chorionic villi derived cells. Molecular analysis can also be done in the presence of a known mutation.
Treatment
Strict lifetime sunlight avoidance is key to reducing the risk of associated sequalae.
Individuals with XP need to take precautions to minimise any sun exposure, including:
- Wearing SPF 50+ and sun-protective clothing such as long sleeves, wide-brimmed hat and wraparound sunglasses.
- Ensuring access to SPF 50+ at all times.
- Avoiding outdoor occupations or recreational activities.
- Avoiding sun exposure through car or building windows, either through drawing curtains and/or applying UV protective tinting to windows.
- Using lightbulbs which emit lower levels of UV.
- Avoiding going outdoors during the middle of the day, and planning travel and activities in periods of low sunlight.
Awareness of your own skin is very important if you suffer from XP, to detect skin cancers early.
It is also worse in people who smoke, and therefore smoking cessation is crucial.
Individuals with XP must also take a vitamin D supplement, as they will be avoiding the sun.
Prevention
Experts recommend genetic counseling for persons with a family history of xeroderma pigmentosum who wish to have children.