Definition
Wells syndrome or Eosinophilic cellulitis is an acute, rare dermatosis of unknown etiology. Eosinophilic cellulitis is reported infrequently in children and is manifested by eosinophil infiltration in the dermis. Eosinophilic cellulitis presents with sudden onset of single or multiple mildly painful or pruritic, erythematous plaques. The lesions are usually seen on the extremities but may be anywhere on the skin and are typically symmetric or widespread. Uncommonly, fever and arthralgias are present. Spontaneous resolution tends to occur over 4-8 weeks. Recurrences may occur. Familial cases have been reported.
Cases may be associated with insect bites, viral infections, parasite infestations, fungal infections, vaccination, and hypersensitivity reactions. More rarely, leukemia, eosinophilic granulomatosis with polyangiitis, and hypereosinophilic syndrome have been reported.
Epidemiology
Wells syndrome (eosinophilic cellulitis) is rare. Only about 80 cases have been reported worldwide. Wells syndrome usually affects adults, but it has been known to occur in children. In one case series of 19 patients, the classic plaque-type presentation was the most common variant found in children, whereas the annular granuloma–like variant was the most common variant in adults.
Pathophysiology of Wells syndrome
The pathogenesis of Wells syndrome is obscure. Many triggering factors have been reported including insect bites, viral infections (parvovirus B19, herpes simplex virus, varicella-zoster virus, mumps virus), parasitic infections (Ascaris, Toxocara Canis, Giardia), bacterial or fungal infections, drugs (antibiotics, non-steroidal anti-inflammatory drugs, thiazide diuretics, anti-TNF, biomedicines) and vaccines. Association of Wells syndrome with other diseases has also been described such as hematologic malignancies (chronic myeloid leukemia, chronic lymphocytic leukemia, polycythemia vera, non-Hodgkin lymphoma), malignant tumors, ulcerative colitis, eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome), hypereosinophilic syndrome).
Wells syndrome may be prior, revealing, or concomitant to these diseases. The fortuitous nature of some of these situations cannot be ruled out, but one must remain vigilant in case of prolonged evolution beyond 6 months, persistent eosinophilia, and/or systemic manifestations associated with Wells syndrome.
The most interesting associations from a pathogenic point of view are certainly those belonging to the spectrum of eosinophilic diseases, such as Shulman syndrome, Churg-Strauss syndrome, and hypereosinophilic syndrome. Wells syndrome could be the first clinical manifestation of these diseases.
The physiopathogenic links between the triggering factors and the associations mentioned above are not clearly established. Inappropriate activation of a Th2-like T lymphocyte clone, synthesizing IL-5, and other eosinophil-stimulating cytokines in response to various, often unidentified, antigenic stimuli is the commonly accepted assumption.
Causes
The cause of eosinophilic cellulitis is unknown. It is believed to be a local hypersensitivity reaction.
Most cases appear to be idiopathic. Implicated triggers or associations include:
- Infection
- Arthropod bite
- Medications and immunizations
- Hematological disorders and malignancies.
Wells syndrome symptoms
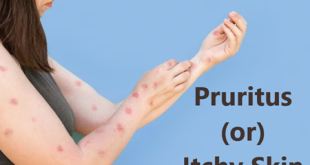
People with Wells syndrome generally develop a skin rash that is often preceded by itching or burning skin.
- The rash consists of raised, red, swollen areas that may be warm to the touch.
- The limbs (arms and legs) are the most commonly affected area of the body. However, the trunk may be involved as well. Symptoms generally come on rapidly and may last for four to eight weeks.
- In some cases, the rash may recur (occur frequently or repeatedly) for years.
- Some people with Wells syndrome may experience symptoms that do not affect the skin, such as asthma, joint pain, fever, or fatigue.
Rash on skin
Complications
Systemic Implications and Complications of Wells syndrome:
Although Wells syndrome typically lacks systemic involvement, some patients can experience malaise and fever. Again, it is imperative to rule out underlying hematologic malignancy and infection in these patients.
Diagnosis
- Skin biopsy is the diagnostic method of choice.
A skin biopsy will show a high number of eosinophils in the skin and underlying tissue, along with patterns of inflammation that are consistent with Wells’ syndrome but not with other possible causes of inflammation such as allergy, parasites, dermatitis, insect bites, or Churg-Strauss syndrome.
- A complete blood count may show a high number of eosinophils.
Wells syndrome treatment
The skin symptoms associated with wells syndrome are typically treated with oral or topical corticosteroids such as Prednisone. Oral corticosteroid treatment with prednisone can lead to a dramatic improvement of eosinophilic cellulitis within days. The course is typically tapered over one month.
Other treatments include minocycline, dapsone, griseofulvin, ciclosporin, and oral antihistamines.
Mild cases may respond to topical steroid therapy alone.
Therapeutic agents
- Corticosteroids: It is the reference treatment for all reactional dermatoses whether neutrophilic or eosinophilic. General corticotherapy reduces the duration and importance of relapses in 10% of cases. Initial doses range from 0.5 to 1 mg/kg/day, with rapid tapering. During the decrease of the treatment, it is not uncommon to observe a relapse of the lesions, which can lead, in certain cases, to a real corticosteroid dependence. Local corticosteroids represent an interesting alternative to general corticosteroids, especially in superficial forms, with inconsistent results of the order of 50%. However, it should not be used in deep hypodermic or extended forms.
- Dapsone: Dapsone is an interesting therapeutic alternative to corticosteroids, with doses ranging from 50 to 200 mg daily. It would reduce the duration of relapses. The optimal duration of treatment is not clearly defined but can be several months in the absence of adverse effects requiring discontinuation of treatment. It can be used alone or in combination with other treatments, including corticosteroids. It can also serve as a relay for general corticosteroids, especially in cases of corticosteroid dependence.
- Antihistamines: Antihistamines, especially hydroxyzine if pruritus, is important and may be tried as a first intention, because of their excellent tolerance even if their effectiveness does not seem to exceed 25% of the cases. They sometimes allow the practitioner to avoid the use of general steroid treatment. The dosage varies from 50 to 100 mg/day. Hydroxyzine can be combined with other antihistamines.
- Other treatments: These are all anecdotal, with their interest being reported in only a few clinical cases. Colchicine, PUVA (psoralen and ultraviolet A) therapy, interferon alpha, cyclins, synthetic antimalarials, ciclosporin, and anti-TNF agents can be mentioned.
Therapeutic strategy
In most cases, general corticosteroids (10 to 80 mg daily) allow rapid healing. Tapering the dose over one month is generally well tolerated. Continued low-dose therapy with corticosteroids allows the prevention of recurrences. Dapsone may be prescribed as a first-line treatment in low-inflammatory forms. It also seems to give good results in the case of corticosteroid resistance. IFN-alpha and IFN-beta could represent interesting alternatives. For mild cases, topical corticosteroids may be sufficient. Finally, the treatment of an associated disease, when it is found, is essential. It must be prescribed as a first-line treatment and can cure Wells syndrome
Prevention of Wells syndrome
- When the body is injured or diseased, the immune system uses a process called inflammation to fight pathogens and facilitate tissue repair and then inhibits the inflammatory response to prevent damage to its own cells and tissues.
- Patients on prolonged bedrest should be given heparin to prevent deep venous thrombosis as well as stool softeners to prevent constipation.
- In addition, some cases suggest that combination therapy with levocetirizine and hydroxyzine may be successfully used as a corticosteroid-sparing treatment or to prevent relapse after the discontinuation of corticosteroid treatment.