


Definition
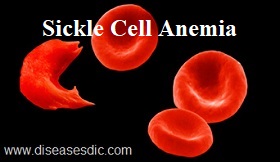
Sickle cell anemia, or sickle cell disease (SCD), is a genetic disease of the red blood cells (RBCs). Normally, RBCs are shaped like discs, which give them the flexibility to travel through even the smallest blood vessels. However, with this disease, the RBCs have an abnormal crescent shape resembling a sickle. This makes them sticky and rigid and prone to getting trapped in small vessels, which blocks blood from reaching different parts of the body. This can cause pain and tissue damage.

Sickle cell and normal blood cell

Normal blood flow and sickle cell blocking blood flow
Types
There are several types of sickle cell disease. The most common are:
- Sickle Cell Anemia (SS),
- Sickle Hemoglobin-C Disease (SC),
- Sickle Beta-Plus Thalassemia,
- Sickle Hemoglobin-D Disease and
- Sickle Beta-Zero Thalassemia.
Sickle Cell Anemia (SS): When a child inherits one substitution beta globin genes (the sickle cell gene) from each parents, the child has Sickle Cell Anemia (SS).
Sickle Hemoglobin- C Disease (SC): Individuals with Sickle Hemoglobin-C Disease (SC) have a slightly different substitution in their beta globin genes that produces both hemoglobin C and hemoglobin S.
Sickle Beta-Plus Thalassemia: Sickle beta-plus thalassemia affects beta globin gene production. The size of the red blood cell is reduced because less beta protein is made. If inherited with the Hb S gene, you will have haemoglobin S beta thalassemia. Symptoms are not as severe.
Sickle Hemoglobin-D Disease: Through research, hemoglobin D, which is a different substitution of the beta globin gene, has been found to interact with the sickle hemoglobin gene.
Sickle Hemoglobin-O Disease: Hemoglobin O, another type of substitution in the beta globin gene, also interacts with sickle hemoglobin.
History
- The symptoms related to sickle cell crises were known by various names in Africa, long before they were recognized in the western hemisphere. Symptoms of sickle cell anemia could be tracked back to year 1670 in one Ghanian family.
- It was in 1910 when James Herrick observed, “peculiar elongated sickle shaped RBCs” in the blood of an anemic black medical student, and then the scientific community came to know about it.
- It was the discovery of Emmel in 1917 of the sickling phenomenon, in vitro, in the members of a family which first suggested the genetic basis for sickling. So it was discovered to be an inheritable condition. Later on it was explained that the sickling phenomenon, in vitro, was due to deprivation of oxygen.
- Both Huck and Sydenstricker, who did the detailed analysis of the pedigrees of Huck’s patients, concluded that the sickle cell phenomenon was inherited as a Mendelian autosomal recessive characteristic.
Statistics on sickle cell anemia
The main pathology of the sickle cell syndrome is an abnormality of the haemoglobin gene, the oxygen carrying protein of the bloodstream. The sickle cell form of this gene is most commonly carried by the African population, with approximately 8% of African Americans carrying the sicle cell disease gene. In other regions such as India and the Middle East, different abnormalities exist in the haemoglobin gene, causing a similar pattern of disease. The gene is relatively uncommon amongst caucasian populations.
Causes of sickle cell anemia
The basic cause of sickle cell anemia involves hemoglobin, a component of the red cells in the blood. The hemoglobin molecules in each red blood cell carry oxygen from the lungs to organs and tissues and then bring back carbon dioxide for removal by the lungs. In sickle cell anemia, this process is disrupted. After the hemoglobin molecules give up their oxygen, some of them may cluster together and form long, rod-like structures that become stiff and assume a sickle shape and gene mutation is also leads to inherited one gene for hemoglobin S from each parent.
Symptoms of sickle cell anemia
- Yellow eyes
- Painful swelling of hands and feet
- Frequent pain episodes
- Stunted growth
- Stroke
- Anemia (looking pale)
- Dark urine
Diagnosis and testing of sickle cell anemia
- A blood test can check for hemoglobin S- the defective form of hemoglobin that underlies sickle cell anemia.
- Sickle cell disease can be diagnosed in an unborn baby by sampling some of the fluid surrounding the baby in the mother’s womb (amniotic fluid) to look for the sickle cell gene.
- DNA analysis: This test is used to investigate alterations and mutations in the genes that produce hemoglobin components. It may be performed to determine whether someone has one or two copies of the Hb S mutation or has two different mutations in hemoglobin genes (e.g., Hb S and Hb C).
Treatment and medications in recent trends
- Rehydration with intravenous fluids helps red blood cells return to a normal state. The red blood cells are more likely to deform and assume the sickle shape if you’re dehydration.
- Treating underlying or associated infections is an important part of managing the crisis, as the stress of an infection can result in a sickle cell crisis. An infection may also result as a complication of a crisis.
- Blood transfusions improve transport of oxygen and nutrients as needed. Packed red cells are removed from donated blood and given to patients.
- Supplemental oxygen is given through a mask. It makes breathing easier and improves oxygen levels in the blood.
- Pain medication like morphine is used to relieve the pain during a sickle crisis.
- Hydroxyurea (Droxia, Hydrea) helps to increase production of fetal hemoglobin. It may reduce the number of blood transfusions.
- Immunizations can help prevent infections. Patients tend to have lower immunity.
- Bone marrow transplant has been used to treat sickle cell anemia.
Prevention of sickle cell anemia
- Drinking plenty of water (hydration).
- Avoiding extremely hot or cold temperatures.
- Avoiding places or situations with low oxygen, such as high altitudes, military boot camp, or strenuous athletic training.
- Getting plenty of rest and taking frequent breaks during exercise.
- Taking the medicine Hydroxyurea. People taking hydroxyurea must be monitored regularly by a doctor to ensure the right dose is given for the best effect.